|
[На
предыдущую главу]
МАТЕРИАЛЫ И МЕТОДЫ
АпоЕ выделяли из изолированных частиц ЛОНП согласно [9]. Восстановление
S–S связей и блокирование SH–групп апоЕ йодацетамидом (алкилирование) проводили,
как описано [10]. Мечение алкилированного апоЕ (5,6 мкМ) дансил-хлоридом
(Serva, ФРГ) или флуоресцеинизотиоционатом (Isomer I, Sigma, США)
проводили в 0,1 М NаНСО3,
рН 9,2 при мольном отношении краска/белок 10:1. Для получения разной стехиометрии
мечения инкубацию проводили в течение разного времени, но не более 14 часов.
Непрореагированную метку отделяли гель-фильтрацией на РD-10 колонке (Pharmacia/LKB,
Швеция), уравновешенной 0,02 М трис-НСl, рН 7,5, 0,15 М NаСl, 0,1 mM ЕDTA
(буфер А), и последующим диализом против 100-кратного избытка буфера
А. Образцы фильтровали через фильтр с диаметром пор 0,45 мкм (HV-тип,
Millipore, США) и концентрацию белка во флуоресцентно-меченных препаратах
определяли по [11] с использованием алкилированного апоЕ в качестве стандарта.
Для нахождения концентрации последнего использовали вычисленный нами из
аминокислотного состава апоЕ коэффициент молярной экстинкции при
280 нм e280
=43376 М–1см–1.
Стехиометрию мечения рассчитывали, принимая во внимание e340
=3400 М–1см–1для
связанного дансила [12] и e495
=42500 М–1см–1
для связанного флуоресцеина [13].
Образцы для флуоресцентных измерений готовили
в буфере А тремя различными способами. Во-первых, растворы дансил-меченного
апоЕ (апоЕ/Д) и флуоресцеин-меченного апоЕ (апоЕ/Ф) в разных соотношениях
донор:акцептор смешивали в объеме 500 мкл (донор-акцепторная пара, образец
ДФ) и инкубировали в течение двух часов при 24°С.
Готовили также три остальных образца, содержащих только апоЕ/Д (только
донор, Д-образец), только апоЕ/Ф (только акцептор, Ф-образец), или только
немеченный апоЕ (контрольный образец). Во всех образцах общую концентрацию
апоЕ сохраняли постоянной и равной 0,6–0,8
мкМ. Во-вторых, четыре различных препарата (см. выше) предварительно денатурировали
4 М Gdn–HCl в течение трех часов при 24°С
и затем диализовали при 4°С
в мешочках для диализа (Serva, ФРГ, предел пропускания 12–14 кДа) с двумя
сменами против 500-кратного избытка буфера А. Третий способ заключался
в солюбилизации четырех образцов в объеме 200 мкл холатом Nа в концентрации
7,5 мМ, что выше критической концентрации мицеллообразования, в течение
двух часов при 24°С
и последующей инкубации в течение ночи при 4°С.
Непосредственно перед диализом препарат разбавляли до 500 мкл буфером А
и диализ проводили в микродиализаторе (Microdialyzer System 500, Pierce,
США) с диализной мембраной Spectra/Рor 4 (предел пропускания 12–14 кДа)
при 24°С
в течение 48 часов с тремя сменами. Диализат представлял собой буфер А
с 0,1 мМ ФМСФ и с 0,5 г адсорбента желчных кислот ХАD-2 (Sigma, США). 100
мл диализата в диализной камере посредством перистальтического насоса постоянно
перемешивались с дополнительными 200 мл раствора в стакане, содержимое
которого также перемешивалось с помощью магнитной мешалки.
Для изучения кинетики процесса ренатурации
апоЕ образцы готовили аналогичным образом, за исключением того, что дансил-
и флуоресцеин-меченный апоЕ смешивали в мольном отношении 1:1, и апобелок
денатурировали или 4 М Gdn–HCl или холатом Na в концентрации 60 мМ. Для
неалкилированных образцов добавляли ДТТ до 10 мМ. После гуанидин- или холат-индуцированной
денатурации образцы диализовали при 4°С
в течение 48 часов с двумя сменами в диализных мешочках против 500-кратного
избытка буфера А с 0,1 мМ ФМСФ и с 0,2 г/л XAD-2. В случае неалкилированных
образцов в буфер добавляли ДТТ до 1 мМ. После диализа один набор проб инкубировали
при 0°С,
другой – при 24°С
и измеряли различные флуоресцентные параметры при 24°С
через различные промежутки времени после снятия препаратов с диализа. Для
измерений в конкретной временной точке каждый раз бралась свежая аликвота
раствора меченого апобелка для исключения фотодеструкции.
Флуоресцентные измерения проводили на спектрофлуориметрах
RF-540 (Shimadzu, Япония) или Aminco-500 (Aminco, США) при 24°С.
Ширина щели на возбуждении была 2 нм, на эмиссии – 4 нм (Aminco-500) или
по 5 нм (RF-540). Из спектров флуоресцентно-меченных образцов вычитали
сигнал из контрольного образца.
Эффективность переноса энергии E
(в %) по сенсибилизации флуоресценции акцептора рассчитывали по формуле:
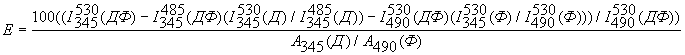 , ,
где в скобках приведено обозначение образца,
I и A
– интенсивности флуоресценции и поглощения, нижние и верхние цифры – длины
волн возбуждения и эмиссии, соответственно. Смысл заключается в вычитании
из сигнала донор-акцепторной пары в области, обогащенной флуоресценцией
акцептора при возбуждении в полосе возбуждения донора, вкладов сигналов
донора и акцептора, непосредственно возбуждаемых при этой длине волны.
Аналогичный прием расчета был использован Borochov-Neori с соавт. [14].
Величину анизотропии флуоресценции r
для флуоресцеин-меченного образца измеряли с учетом G-фактора,
корректирующего поляризацию в монохроматоре эмиссии [15]. Все результаты
измерений корректировали с учетом светорассеяния. Длины волн возбуждения
и регистрации флуоресценции составляли, соответственно, 490 и 520 нм. Для
тушения флуоресценции апоЕ/Ф анионами I–
долю доступных флуорофоров f
и константу тушения Штерн-Фольмера КШ-Ф определяли при
экстраполяции модифицированной зависимости Штерн-Фольмера [15]:
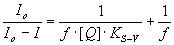 , ,
где Io
и I – интенсивность флуоресценции
в отсутствии и в присутствии тушителя, на область бесконечно больших концентраций
анионов I–.
“Сшивание” апоЕ в концентрации 0,72 мкМ
проводили в буфере Б водорастворимым бифункциональным реагентом
ЭДК в концентрации 20 мМ при 24°С.
В разные промежутки времени отбирали аликвоты и реакцию останавливали добавлением
буфера для образцов для электрофореза в системе с ДДС–Na [16]. После разделения
продуктов реакции электрофорезом в ПААГ в градиенте концентраций акриламида
5–20% в системе с ДДС–Na гель окрашивали серебром [17].
[На
следующую главу] [На
оглавление] [На
список сокращений и обозначений]
Copyright ©
|