Extended abstract for "18th Symposium on Halogenated Environmental Organic Pollutants Organohalogen Compounds - Dioxin'98", Stockholm, Sweden, August 17 - 21, 1998, in Organohalogen Compounds, 36, pp 97-100.
Thermodynamic functions of dibenzo-p-dioxin and its polychlorinated derivatives in the gaseous and condensed phases
Vladimir S. Iorish and Olga V. Dorofeeva
Glushko Thermocenter of Russian Academy of Sciences, "IVTAN" Association of RAS, HEDRC, Izhorskaya 13/19, Moscow 127412, Russia
Introduction
The availability of thermodynamic data for polychlorinated dibenzo-p-dioxins is of fundamental importance for understanding of the mechanism for their formation in order to assist in design of strategies to effectively control or eliminate their emission. Last decade considerable progress had place in thermodynamic properties study of these compounds owing to experimental and theoretical investigations[1-8]. However, thermodynamic database creating requires to develope a data set which is consistent with all available information, general laws of thermodynamics and some correlations.
The aim of this work is to combine for the first time the data for gaseous and condensed phases to derive such consistent data set.
Material and Methods
There are no experimental data on entropies and heat capacities of gaseous dibenzo-p-dioxin (DD) and polychlorinated dibenzo-p-dioxins (PCDDs) and their values were estimated by group additivity approach and by statistical thermodynamics method [1,4,5]. The S o(T) and 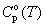 values evaluated in these works differ from one another by 10 - 60 J.K-1.mol-1. Recent ab initio calculation of vibrational spectra of four tetrachlorodibenzo-p-dioxins [8] makes it possible to predict the values of entropy and heat capacity of PCDDs more reliably.
values evaluated in these works differ from one another by 10 - 60 J.K-1.mol-1. Recent ab initio calculation of vibrational spectra of four tetrachlorodibenzo-p-dioxins [8] makes it possible to predict the values of entropy and heat capacity of PCDDs more reliably.
Structural parameters and vibrational frequencies needed for statistical thermodynamics calculation were estimated in this work using the similarity transference procedure. Based on X-ray diffraction and theoretical data for DD and some of PCDDs, the structural parameters were evaluated to be the same for all PCDDs. Available vibrational assignments for dibenzofuran(DF), PCDDs as well as for chlorinated benzenes, anthracene, 1,4-dioxin, and furan were used in this work to develop the simplified force field approximation for PCDDs. The distinguishing feature of proposed method is that the force constants are calculated to best fit not only the vibrational frequencies, but also the experimental data on entropy or heat capacity if the later are known in the literature. Twenty-six force constants were obtained by a least-squares refinement to provide a satisfactory fit to known fundamentals of DF, PCDDs, chlorinated benzenes and to the experimental values of entropy of DF. Using estimated structural parameters and vibrational frequencies, the thermodynamic functions of 75 possible isomers of PCDDs were calculated in this work by rigid-rotor harmonic-oscillator approximation.
The only experimantal information on thermodynamic functions of PCDDs in condensed phase is indirectly contained in several vapor pressure measurements for solid [2] and overcooled liquid [3] compounds. To extract the data on thermodynamic functions from these experimental results we should make some assumptions on heat capacity of solid and liquid phases. Domalsky and Hearing[9] showed the possibility of satisfactory estimation of the heat capacity for solid organic compounds at 298.15K by group additivity method. In the absence of all required increments for the heat capacity of PCDDs estimation, we used "difference method", which is fully consistent with the group additivity approach. Using experimental heat capacity values for several related compounds we estimated Cpo(DD,s, 298.15) = 215  5 J.
K-1.
mol-1. From this value and increments for chlorine substitutions developed by Domalski and Hearing[9] heat capacities for all solid PCDDs were estimated by equations:
5 J.
K-1.
mol-1. From this value and increments for chlorine substitutions developed by Domalski and Hearing[9] heat capacities for all solid PCDDs were estimated by equations:
Cpo(xCl-DD) = Cpo(DD) + x*( Cpo[CB-(Cl)(CB)2] - Cpo[CB-(H)(CB)2]), (1)
where Cpo[CB-(Cl)(CB)2] = 33.55 J.
K-1.
mol-1, Cpo[CB-(H)(CB)2] = 20.13 J.
K-1.
mol-1
To extrapolate the heat capacity from room temperature up to the melting point we used the linear equation derived from experimental data for related compounds:
Cpo(T)/R = Cpo(298.15)/R + 1.51.
n.
(T � 298.15)/Tm, (2)
where n is number of atoms in the molecule, Tm is the melting temperature, and R is gas constant.
Bondi-Rowlinson equation[10] and the heat capacities for gases calculated in this work were used for heat capacity estimation of liquid PCDDs :
CpLo /R= CpGo/R + 2.56 + 0.436.
(1 � Tr)-1 + w.
[2.91 + 4.28.
(1 � Tr)1/3Tr-1
+ 0.296.
(1 � Tr)-1] (3)
where Tr = T/Tc , Tc is the critical temperature,  is the acentric factor. Tc values were calculated by group contribution technique [11]. Boiling points required for these calculations were obtained from full set of thermodynamic data for gaseous and condensed phases (see Table 2). So several iterations were needed to agree the data. The final heat capacities were approximated by linear dependence on temperature (see below, Table 2).
is the acentric factor. Tc values were calculated by group contribution technique [11]. Boiling points required for these calculations were obtained from full set of thermodynamic data for gaseous and condensed phases (see Table 2). So several iterations were needed to agree the data. The final heat capacities were approximated by linear dependence on temperature (see below, Table 2).
Vapor pressure data for several PCDDs [2] were treated by "Third Law" and "Second Law" techniques taking into account direct determination the enthalpy of sublimation for some of PCDDs[6,7]. As a result of these treatments standard entropy values and enthalpies of sublimation were estimated for the considered solid PCDDs. Rordorf's procedure [2] for enthalpies of fusion estimation is used for the cases where the values were not known from experiment.
Results and Discussion
To approximate calculated thermodynamic properties of gases, we develop the group additivity scheme with ten parameters (Table 1). According to designation by Benson [12] groups A and B can be written as CB�(H) and CB�(Cl), respectively. The group D consists of two O�(CB)2 and four CB�(CB)2(O) groups and describes the dioxin frame taken as a whole. Six  corrections are applied for 1,2-, 1,3-, 1,4-, 1,2,3-, 1,2,4- and 1,2,3,4- interactions of chlorine atoms. For example, the equation for calculating the entropy of 2,3,7,8-TCDD contains the following groups:
corrections are applied for 1,2-, 1,3-, 1,4-, 1,2,3-, 1,2,4- and 1,2,3,4- interactions of chlorine atoms. For example, the equation for calculating the entropy of 2,3,7,8-TCDD contains the following groups:
S o(2,3,7,8-T4CDD) = D + 4A + 4B +2
12 = 510.5 kJ.
mol-1
TABLE 1. Group additivity values for gaseous DD and PCDDs
|
|
|
|
|
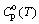 |
|
Group |
|
|
|
298.15 |
300 |
400 |
| |
kJ.mol-1 |
J.K-1.mol-1 |
|
A |
13.765 |
3.2568 |
48.327 |
21.032 |
21.170 |
28.188 |
|
B |
-16.835 |
6.5248 |
77.662 |
36.422 |
36.556 |
42.891 |
|
D |
-169.32 |
2.1373 |
20.764 |
11.788 |
11.825 |
13.556 |
|
12 |
8.8 |
0.03666 |
-1.3300 |
0.10790 |
0.10581 |
0.026337 |
|
13 |
4.3 |
0.092512 |
-0.012373 |
0.17136 |
0.16809 |
0.062259 |
|
14 |
1.1 |
0.25399 |
0.50427 |
0.75425 |
0.74864 |
0.50288 |
|
123 |
13.0 |
0.21565 |
-2.4838 |
0.51584 |
0.50735 |
0.19421 |
|
124 |
1.1 |
0.34388 |
-1.0075 |
0.92063 |
0.91052 |
0.50851 |
|
1234 |
14.4 |
0.52376 |
-3.4338 |
1.4004 |
1.3818 |
0.67353 |
TABLE 1 � continued
| |
|
|
Group |
500 |
600 |
800 |
1000 |
1200 |
1400 |
1500 |
| |
J.K-1.mol-1 |
|
A |
34.028 |
38.685 |
45.383 |
49.858 |
52.985 |
55.237 |
56.128 |
|
B |
47.620 |
51.115 |
55.636 |
58.229 |
59.810 |
60.831 |
61.208 |
|
D |
14.882 |
15.909 |
17.326 |
18.202 |
18.770 |
19.155 |
19.302 |
|
12 |
-0.0090665 |
-0.022808 |
-0.026440 |
-0.022413 |
-0.018044 |
-0.01444 |
-0.012981 |
|
13 |
0.027975 |
0.015630 |
0.0080352 |
0.0053498 |
0.0039564 |
0.0029721 |
0.0026854 |
|
14 |
0.35128 |
0.25802 |
0.15688 |
0.10617 |
0.076696 |
0.05805 |
0.051180 |
|
123 |
0.058268 |
0.0007798 |
-0.031310 |
-0.032610 |
-0.028221 |
-0.02356 |
-0.021351 |
|
124 |
0.30360 |
0.19578 |
0.098823 |
0.059467 |
0.039951 |
0.02854 |
0.024892 |
|
1234 |
0.33468 |
0.17140 |
0.046691 |
0.010275 |
-.0016937 |
-0.005727 |
-.0061494 |
Estimation of enthalpy of formation is described in these Abstracts (see V.P. Kolesov et al.)
a
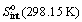 =
= 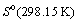 + R ln
+ R ln  , where
is the symmetry number.
, where
is the symmetry number.
The set of thermodynamic data derived for solid and liquid DD and several PCDDs is presented in Table 2 (The enthalpies of formation presented in the table are based on experimental data and estimates for gases). These thermodynamic functions were correlated with chlorine substitution (x) and the following equations allow estimate the same data for all other PCDDs:
So(298.15) = 214.8 + 23.02 x J.
K-1.
mol-1 ,
DsHo(298.15) = 91.6 + 7.93 x kJ.
mol-1 ,
DmHo = 15.7 + 5.37 x kJ.
mol-1 ,
aL = 149.9 +31.42 x J.
K-1.
mol-1 ,
bL = 3971 - 292.0 x J.
mol-1.
10000,
where aL and bL are coefficients in the heat capacity equation (Cpo(T) = aL + bLT/10000) for liquid PCDDs. Eq.(2) should be added to these correlations to estimate thermodynamic functions for all PCDDs with known melting points. For PCDDs with unknown melting points symmetry dependent correlation of fusion temperatures with moments of inertia of the molecules is used.
TABLE 2. Estimated thermodynamic properties of DD and PCDDs in solid and liquid states
|
Substance |
DsHo |
DfHo |
So(298.15) |
Tm |
DmH |
aS |
bS |
aL |
bL |
Tb |
|
at 298.15, kJ.
mol-1 |
J.
K-1.
mol-1 |
K |
kJ.
mol-1 |
Cp(T)=a+bT/10000 J.
K-1.
mol-1 |
o C |
|
DD |
90.8 |
-148.7 |
211.5 |
392.5 |
21.9 |
5.16 |
7038.0 |
143.5 |
4127 |
296 |
|
1-MCDD |
97.0 |
-183.4 |
243.6 |
374.0 |
20.0 |
8.28 |
7386.2 |
171.8 |
3853 |
317 |
|
2-MCDD |
97.8 |
-187.6 |
238.3 |
360.8 |
22.1 |
0.22 |
7656.5 |
178.9 |
3723 |
327 |
|
2,3-DCDD |
108.9 |
-220.5 |
255.4 |
431.6 |
27.1 |
51.20 |
6399.6 |
211.1 |
3411 |
352 |
|
2,7-DCDD |
106.9 |
-227.3 |
272.2 |
482.7 |
28.0 |
71.38 |
5722.7 |
225.8 |
3165 |
384 |
|
2,8-DCDD |
110.1 |
-230.5 |
259.4 |
424.2 |
26.6 |
47.84 |
6512.0 |
219.4 |
3265 |
396 |
|
1,3,7-T3CDD |
117.4 |
-264.1 |
282.0 |
421.7 |
30.8 |
60.19 |
6550.7 |
247.3 |
3021 |
410 |
|
1,2,4-T3CDD |
115.6 |
-265.5 |
276.9 |
401.7 |
31.5 |
50.47 |
6876.8 |
239.8 |
3152 |
393 |
|
1,2,3,4-T4CDD |
120.4 |
-287.6 |
307.6 |
462.2 |
33.0 |
90.81 |
5976.6 |
278.4 |
2751 |
427 |
|
2,3,7,8-T4CDD |
127.7 |
-291.7 |
312.0 |
578.2 |
38.9 |
126.56 |
4777.5 |
274.4 |
2830 |
443 |
|
OCDD |
153.3 |
-428.5 |
397.8 |
604.2 |
59.6 |
186.69 |
4571.9 |
384.0 |
1965 |
502 |
Acknowledgements
This research was supported by Russian Foundation for Basic Research (RFBR), Grant 96-02-016223.
References
- Shaub, W. M.; Thermochim. Acta 1982, 55, 59.
- Rordorf, B.F.;Chemosphere 1989, 18, 783.
- Eltzer, B.D., and Hits, R.A.;Environ.Sci.Technol. 1988, 22, 1362.
- Thompson, D.; Thermochim. Acta 1995, 261, 7.
- Dorofeeva, O.V. and Gurvich, L.V.; Zh. Fiz. Khim. 1996, 70, 7.
- Papina, T. S., Kolesov, V.P., Vorobieva, V.P., and Golovkov, V.F.; J. Chem. Thermodynamics 1996, 28, 307.
- Luk'yanova, V.A., Kolesov, V.P., Avramenko, N.V., Vorobieva, V.P., and Golovkov,V.F.; Russian J.Phys.Chem. 1997, 71, 338.
- Rauhut, G., Pulay, R.; J.Am.Chem Soc. 1995, 117, 4167.
- Domalski, E. S., and Hearing, E. D.; J.Phys. Chem. Ref. Data 1993, 22, 805.
- Reid, R. C., Prausnitz, J. M., and Sherwood, T. K.; The properties of gases and liquids. Third edition. Chapters 2 and 5, McGraw-Hill Book Company, New York , 1977.
- Somayajulu, G. R.;J.Chem.Eng.Data. 1989, 34, 106.
- Benson, S.W.; Thermochemical Kinetics, Wiley, New York, 1976.