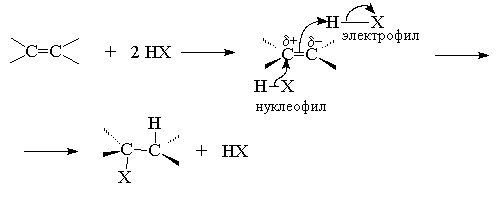
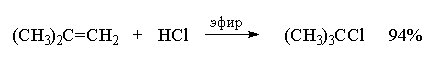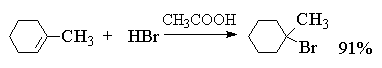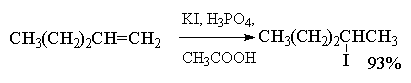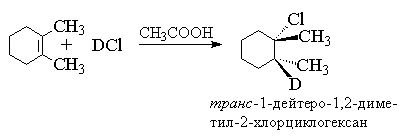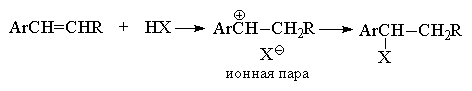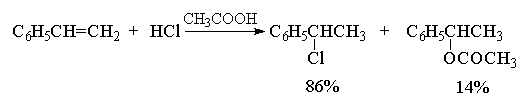4.3.б. Присоединение галогеноводородов
(гидрогалогенирование)
Другой важной реакций электрофильного
присоединения к алкенам является давно
известное гадрогалогенирование алкенов.
Ниже приведены типичные примеры присоединения
HCl, HBr и HI к различным алкенам.
Влияние алкильных заместителей у двойной связи
на скорость присоединения описывается следующей
последовательностью:
Это согласуется с таким механизмом, в котором в
определяющей скорость стадии реакции происходит
образование карбокатиона, поскольку
стабильность алкильных катионов убывает в ряду
третичный > вторичный > первичный.
Таким образом, механизм присоединения должен
включать промежуточное образование или
свободного карбокатиона, что наблюдается редко,
или интермедиата с карбокатионным характером.
Последний случай наиболее типичен.
Если бы присоединение происходило через
"свободный карбокатион", то реакция была бы
совершенно нестереоселективной, так, как
алкильные катионы имеют плоское строение.
Однако, гидрогалогенирование, как правило,
протекает стереоселективно, причем в
зависимости от типа алкена может наблюдаться
селективное анти-присоединение, селективное син-
или смешанное син-анти-присоединение.
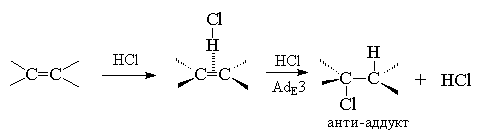
Для алкенов, у которых двойная связь не
сопряжена с ароматическим кольцом, характерно анти-присоединение
галогеноводорода. Анти-присоединение
хлористого и бромистого водорода, хлористого и
бромистого дейтерия наблюдается для
циклогексена, циклопентена, 1,2-диметилгексена,
1,2-диметилпентена, цис- и транс-бутена-2,
гексена-3 и многих других простых алкенов и
циклоалкенов.
В продукте присоединения одинаковые
заместители (метильные группы) расположены по
разные стороны средней плоскости
циклогексанового кольца, следовательно он
относится к транс-ряду. Анти-присоединение
трудно совместимо с механизмом, в котором
предполагается образование дискретного
карбокатиона. Для плоского карбокатиона
нуклеофильная атака галогенид-иона
равновероятна с обеих сторон плоскости, что
должно привести к образованию смеси продуктов син-
и анти-присоединения. Кинетика
гидрогалогенирования алкенов также указывает на
более сложный механизм присоединения. Для
несопряженных алкенов скорость реакции
описывается уравнением третьего порядка со
вторым порядком по галогеноводороду, т. е.
соответствует AdE3-механизму.
Анти-присоединение и второй порядок реакции
по галогеноводороду хорошо согласуется с AdE3-механизмом,
в котором алкен взаимодействует с двумя
молекулами галогеноводорода, одна из которых
выполняет функцию электрофильного, а другая -
нуклеофильного агента.
Такой тримолекулярный механизм предполагает,
что первоначально образуется комплекс алкена и
одной молекулой галогеноводорода с последующей
атакой второй молекулы НХ на этот комплекс с анти-стороны
без образования дискретного карбокатиона.
Следует особо отметить, что любой
тримолекулярный механизм должен состоять из
двух последовательных стадий, поскольку
одновременно столкновение трех молекул крайне
маловероятно.
Анти-присоединение свидетельствует о
предпочтительной нуклеофильной атаке
галогеноводорода со стороны противоположной
той, откуда происходит протонирование алкена.
Вместо галогеноводорода функцию нуклеофильного
агента в конечной стадии может выполнить и
галогенид-ион. Действительно, скорость реакции
обычно возрастает прямо пропорционально
концентрации галогенид-иона, введенного в
реакционную смесь в виде галогенидов
тетраалкиламмония NR4+ X- или лития
LiX. В этом случае наблюдается
стереоспецифическое анти-присоединение.
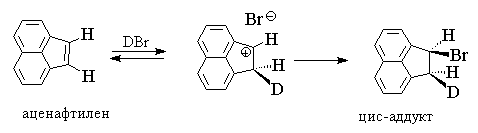
Для алканов, у которых двойная связь сопряжена
с ароматическим кольцом, характерно син-присоединение
или смешанное син-анти-присоединение
галогеноводорода, например:
Нельзя ожидать, что механизм присоединения с
участием ионных пар будет отличаться высокой
стереоселективностью. Если ионная пара
превращается в конечный продукт быстрее, чем
происходит вращение вокруг простой
углерод-углеродной связи, конечным результатом
будет син-присоединение, где протон и
галогенид-ион присоединяется с одной и той же
стороны двойной связи. В противном случае
наблюдается образование продуктов как син-
так и анти-присоединения НХ. Такой случай
реализуется при гидрогалогенировании пара-замещенных
стиролов Z-C6H4-CH=CH2. Наблюдаемая
здесь закономерность заключается в том, что син-присоединение
характерно лишь для тех олефинов, которые при
протонировании дают относительно стабильный
карбокатион, т. е. в случае донорных
заместителей Z.
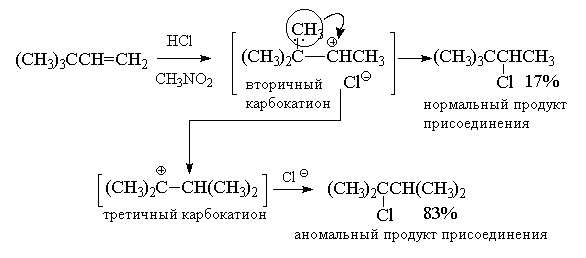
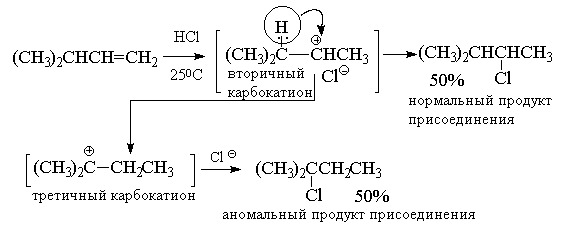
В качестве примера перегруппировок с
1,2-миграцией алкильной группы и гидрид-иона
приведем реакции гидрогалогенирования
соответственно трет-бутилэтилена и
изопропилэтилена.
4.3.в. Ориентация. Правило Марковникова
В отличие от симметричных электрофилов (Hal2),
галогеноводороды представляют собой
несимметричные электрофильные реагенты.
Присоединение любого несимметричного
электрофила (HBr, ICl, H2O, Hg(OAc)2 и т. д.) к
несимметричному алкену в принципе могло бы дать
смесь двух альтернативных продуктов, однако на
практике обычно образуется только один из них. В
качестве примера рассмотрим присоединение
бромистого водорода к пропилену.
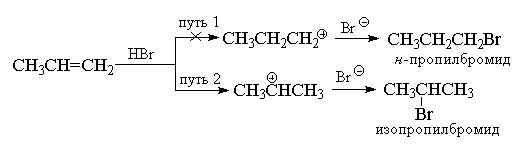
Еще в 1870 г. В.В. Марковников сформулировал
эмпирическое правило, согласно которому
несимметричные алкены присоединяют НХ таким
путем, что преимущественно образуется продукт, в
котором водород присоединяется к наименее
замещенному, а Х - к наиболее замещенному концу
двойной связи.
Обычно правило Марковникова объясняют
различием в стабильности двух альтернативных
карбокатионов. Например, в приведенном выше
примере нормальный н-пропильный катион
значительно менее стабилен, чем изопропильный
катион, и поэтому реакция идет по второму пути.
Правило Марковникова первоначально
использовалось только для случаев присоединения
НХ к углеводородным субстратам, но в принципе его
можно распространить и на реакции других
замещенных алкенов. Так, присоединение НCl к CF3CH=CH2
дает "анти-марковниковский" продукт CF3CH2CH2Cl.
Этого и следовало ожидать, поскольку катион CF3CH+
CH3 менее стабилен, чем катион CF3CH2CH2+
из-за сильного (-I)-эффекта CF3-группы.
Преимущественно образуется катион CF3CH2CH2+
, но он тоже, хотя и в меньшей степени
дестабилизирован индуктивным эффектом CF3-группы,
вследствие чего присоединение HCl к
трифторметилэтилену идет значительно медленнее,
чем присоединение к незамещенному этилену.
По аналогичной причине катионы
винилтриалкиламмония присоединяют HBr также
против правила Марковникова:
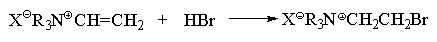
Присоединение НХ к алкенам, имеющим сильные (-I)
и (-M)-заместители, например, к акрилонитрилу или
нитроэтилену также должно идти против правила
Марковникова. Однако в этом случае двойная связь
настолько сильно дезактивирована по отношению к
электрофильным реагентам, что эти реакции идут
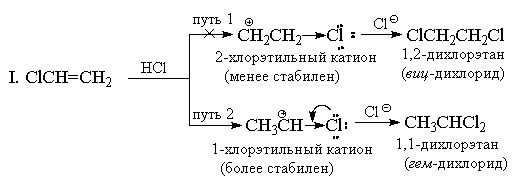
лишь в очень жестких условиях. Хлористый винил СН2=СНСl
всегда дает исключительно "марковниковские
аддукты". Например, при его реакции с HCl
образуется только 1,1-дихлорэтан (геминальный
дихлорид) CH3CHCl2. Хлор, аналогично CF3-группе
имеет сильный (-I)-эффект, и на первый взгляд,
кажется, что по этой причине присоединение
должно иметь антимарковниковскую ориентацию,
т. к. катион +CH2CH2Cl должен быть
более стабильным, чем катион СН3СН+Cl.
Однако, в отличие от CF3-группы, хлор кроме
(-I)-эффекта обладает также противодействующим
ему (+М)-эффектом (т. к. имеет неподеленные пары).
Опыт показывает, что величина мезомерного
эффекта вполне достаточна, чтобы понизить
энергию 1-хлорэтильного катиона ниже уровня
энергии 2-хлорэтильного катиона, в котором
+М-эффект не проявляется.
II. С позиций теории резонанса строение
1-хлорэтильного катиона может быть представлено
следующим образом:
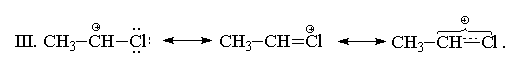
Тем не менее, присоединение к хлористому винилу
происходит медленнее, чем к этилену в тех же
условиях, т. е. по суммарному эффекту
(-I > +M) хлор остается электроноакцепторным
заместителем по сравнению с водородом, а
1-хлорэтильный катион менее стабилен, чем С2Н5+.
Аналогичным образом реагируют с НХ и другие
винилгалогениды.
Виниловые эфиры CH2=CHOR присоединяют НХ (X=Hal)
по правилу Марковникова с гораздо большей
скоростью, чем все перечисленные выше замещенные
алкены. Это связано со значительным +М-эффектом
RО-группы. В отличие от атома хлора, RО-группа по
суммарному электронному эффекту (+М > -I)
является сильным электронодонорным
заместителем, эффективно стабилизирующим
соседний карбокатионный центр. Строение
карбокатиона в этом случае также может быть
представлено в виде набора двух резонансных
структур
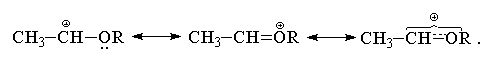
Атака оксониевого катиона галогенид-анионом
приводит к образованию  -галогенэфиров
типа СН3СН(Hal)OR.
-галогенэфиров
типа СН3СН(Hal)OR.
4.3.г. Гидратация алкенов
Кислотно-катализируемая гидратация алкенов
приводит к образованию спиртов. Направление
гидратации алкенов определяется правилом
Марковникова, поэтому предполагается, что в
качестве промежуточной частицы в этой реакции
образуется карбокатион.
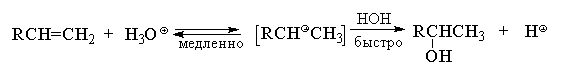
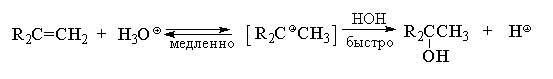
Склонность вторичных алкильных карбокатионов
к перегруппировкам мешает использованию
гидратации алкенов для получения вторичных
спиртов.
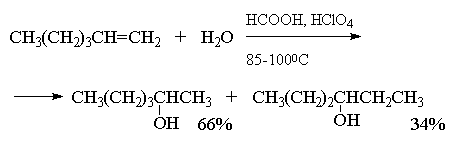
Этот метод в лаборатории нашел ограниченную
область применения только для получения
третичных спиртов. Реакция гидратации в этом
случае в значительной степени обратима и
третичные спирты образуются с низкими выходами
(40-45%).
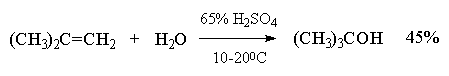
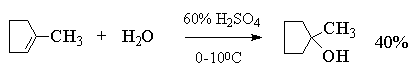
Гидратация простейших алкенов - этилена и
пропилена - представляет собой важный
промышленный метод получения этилового и
изопропилового спиртов.
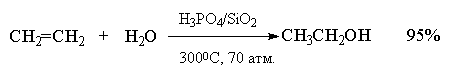
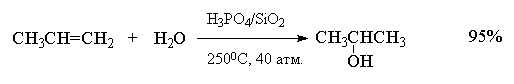
В лабораторной практике прямая гидратация
алкенов не нашла широкого применения как
вследствие жестких условий, так и благодаря
образованию значительного количества изомерных
спиртов. В настоящее время для региоселективного
получения спиртов из алкенов обычно
используется родственная реакция
гидроксимеркурирования - демеркурирования.
4.3.д.
Гидроксимеркурирование-демеркурирование
Электрофильная атака на двойную связь алкена
может осуществляться ионами металлов, среди
которых особое положение занимает катион ртути
(II). Ацетат ртути в очень мягких условиях при 200С
присоединяется к алкенам в водном
тетрагидрофуране (ТГФ) или в водной уксусной
кислоте с образованием ртутьорганических
соединений. Присоединение ацетата ртути по
двойной связи протекает региоспецифично в
строгом соответствии с правилом Марковникова,
т. е. катион ртути присоединяется к наименее
замещенному атому углерода.
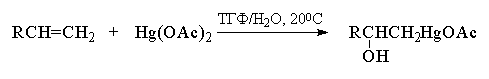
Связь С-Hg в ртутьорганических соединениях
может быть легко расщеплена под действием
боргидрида натрия NaBH4, с образованием ртути
и новой связи С-Н. Предполагается, что в качестве
нестабильного интермедиата при этом получается
алкилмеркургидрид, который далее разлагается с
выделением металлической ртути по радикальному
механизму.
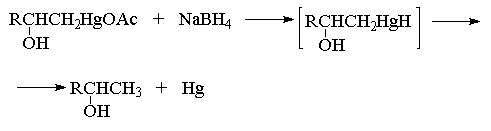
Суммарно этот двухстадийный процесс
гидроксимеркурирования-демеркурирования в
конечном итоге представляет собой
региоспецифичную гидратацию алкена по правилу
Марковникова в исключительно мягких условиях,
когда образование побочных продуктов сведено к
предельно возможному минимуму. Это можно
наглядно проиллюстрировать с помощью следующих
примеров, в которых суммарный выход продуктов
реакции составляет 90-98%. Приведенные цифровые
данные в этом случае обозначают не выходы
образующихся соединений, а их соотношение в
смеси.
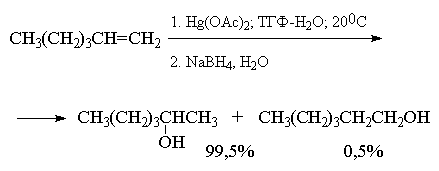
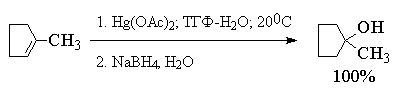
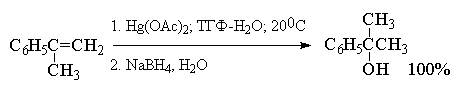
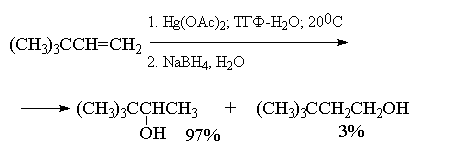
Как видно из приведенных выше примеров
гидроксимеркурирование-демеркурирование
алкенов в большинстве случаев обеспечивает
региоспецифическую гидратацию алкенов с
образованием практически только одного из двух
изомерных спиртов. Следует отметить, что нет
никакой необходимости в выделении
ртутьорганического соединения, и оба процесса
могут быть проведены непосредственно один за
другим.
Гидроксимеркурирование несимметричных
алкенов, по-видимому, начинается с атаки катиона
AcOHg+ и образования в качестве интермедиата
несимметричного циклического меркуриниевого
катиона (аналога несимметричного
галогенониевого иона), который затем
раскрывается в результате нуклеофильной атаки
водой по наиболее замещенному атому углерода,
несущему больший положительный заряд.
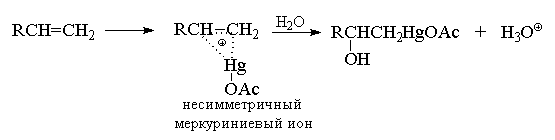
Мостиковый меркуриниевый ион можно
зафиксировать в ненуклеофильной сильнокислой
среде даже при 200С при присоединении более
сильного электрофильного агента -
трифторацетата ртути в смеси фторсульфоновой
кислоты и пятифтористой сурьмы.
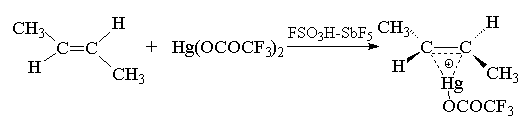
Меркуриниевый катион может расщепляться не
только при действии воды, но и других
электронодонорных растворителей: спиртов,
уксусной кислоты, ацетонитрила и др. Конечным
продуктом реакции в этом случае будут
соответственно простые эфиры, ацетаты или
N-замещенные амиды уксусной кислоты, например:
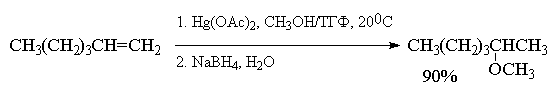
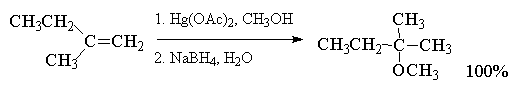
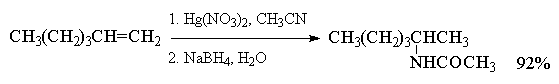
При использовании в реакции
алкоксимеркурирования-демеркурирования
разветвленных вторичных или третичных спиртов
более эффективными, чем ацетат ртути, являются
трифторацетат Hg(OCOCF3)2 или трифлат
ртути Hg(OSO2CF3)2.

Таким образом, гидрокси- и
алкоксимеркурирование-демеркурирование - это
один из лучших препаративных методов синтеза
спиртов и простых эфиров с разветвленными
алкильными радикалами.
Присоединение солей ртути к алкенам
представляет собой наиболее яркий пример
реакции сопряженного присоединения к двойной
связи, где роль внешнего нуклеофильного агента
выполняет растворитель. Стереохимия двойного
процесса
гидроксимеркурирования-демеркурирования
зависит от стереохимического результата каждой
отдельной стадии. Для гидроксимеркурирования
характерно анти-присоединение, как и для
других реакций с участием циклического катиона.
Однако радикальное демеркурирование не
отличается высокой стереоселективностью.
Поэтому весь процесс в целом также
нестереоспецифичен.
4.3.е. Присоединение алкенов (катионная
димеризация и полимеризация алкенов)
Наиболее интересным примером такого типа
реакций является димеризация и полимеризация
изобутилена в присутствии серной кислоты.
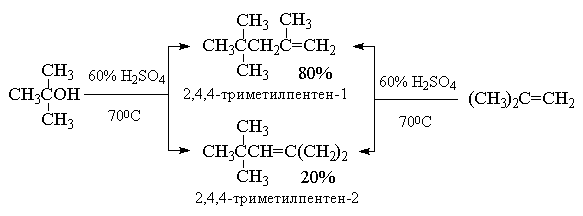
Техническое название этой смеси алкенов -
"диизобутилен". Данная реакция протекает по
катионному механизму, сходному с механизмом
присоединения минеральных кислот к двойной
связи алкенов. На первой стадии протон
присоединяется к молекуле изобутилена с
образованием относительно стабильного трет-бутилкатиона.
Далее образовавшийся трет-бутилкатион
(кислота Льюиса) реагирует с молекулой
изобутилена (основание Льюиса) с образованием
нового стабильного третичного октильного
катиона.
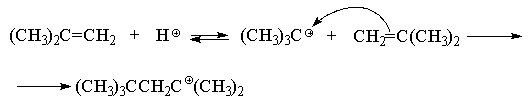
В данных условиях под действием оснований (H2O,
HSO4--ионы) октильный карбокатион
быстро теряет протон и превращается в смесь
изомерных пентенов, т. к. отрыв протона
происходит из двух различных положений:
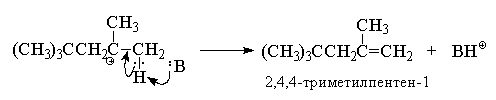
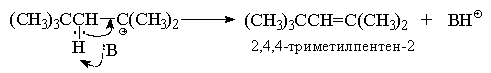
Преимущественное образование
термодинамически менее стабильного алкена -
2,4,4-триметилпентена-1 (80% в реакционной смеси )
связано с большей пространственной доступностью
для атаки основанием атомов водорода метильных
групп, по сравнению с атомами водорода
метиленовой группы. В промышленности
"диизобутилен" гидрируют на Ni-Ренея и
получают "изооктан" (техническое название
углеводорода 2,2,4-триметилпентана), используемый
в качестве высокооктанового топлива для
двигателей внутреннего сгорания.
При высоких концентрациях серной кислоты
(более 80%) происходит катионная полимеризация
изобутилена с образованием полимера,
называемого полиизобутиленом (-CH2C(CH3)2-)n.
Этот каучукоподобный полимер используют для
получения антикоррозионных и гидроизоляционных
покрытий, герметиков и др.
Кроме изобутилена, по катионному механизму
полимеризуются 3-метилбутен-1, виниловые эфиры и
некоторые производные стирола, способные
образовывать сравнительно устойчивые
карбокатионы. В качестве катализаторов
катионной полимеризации также используют
фтористый водород и кислоты Льюиса: BF3, AlCl3,
AlBr3 и др. в присутствии очень малых
количеств воды.
4.3.ж. Присоединение алканов
(алкилирование алкенов)
Другой промышленный метод синтеза
"изооктана" основан на взаимодействии
изобутилена с избытком изобутана в присутствии
концентрированной серной кислоты или в
безводном фтористом водороде при 0 100С.
100С.
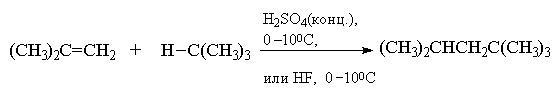
Эта реакция также протекает по катионному
механизму и, что особенно интересно, является
примером цепного катионного процесса. Сначала
изобутилен в условиях реакции димеризуется с
образованием третичного октильного катиона (CH3)3CCH2C+(CH3)2.
Подробно механизм его образования изложен в
предыдущем разделе. Далее происходит быстрый
перенос водорода (в виде гидрид-иона) от
изобутана к октильному катиону с образованием
молекулы "изооктана" и нового
трет-бутилкатиона, который в свою очередь быстро
реагирует с изобутиленом с образованием нового
октильного катиона и т. д.
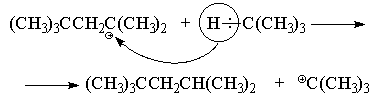
Кроме синтеза "изооктана", такой метод
алкилирования используется в нефтехимической
промышленности для синтеза высококипящих
разветвленных углеводородов из разветвленных
алкенов и алканов низкокипящих фракцией
термического крекинга.